I’m trying to download some data from the Sequence Read Archive but it seems to be behaving quite unusually. Just writing a post to summarise this and see if anyone has seen it before. If I get an answer from the SRA helpdesk, I will post it in the comments/an edit.
I’m interested in ERR024630, or here is the ENA version. When I use the SRA toolkit to download the data
`sratoolkit.2.5.2-centos_linux64/bin/fastq-dump –split-files ERR024630`
This, somewhat confusingly results in 3 fastq files.
-rw-r–r– 1 philip philip 444M Jan 4 17:30 ERR024630_1.fastq
-rw-r–r– 1 philip philip 180M Jan 4 17:30 ERR024630_3.fastq
-rw-r–r– 1 philip philip 425M Jan 4 17:30 ERR024630_4.fastq
_3 only has one base per read, but the same number of lines as _1 and _4
Weird huh!?
So, I downloaded the same data from the ENA, just using
`wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR024/ERR024630/ERR024630_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR024/ERR024630/ERR024630_2.fastq.gz`
Ok, now the real weirdness starts. These files have the same number of lines as the ones downloaded from SRA. They have the same read headers, and the corresponding reads share a large overlap, but are not 100% identical.
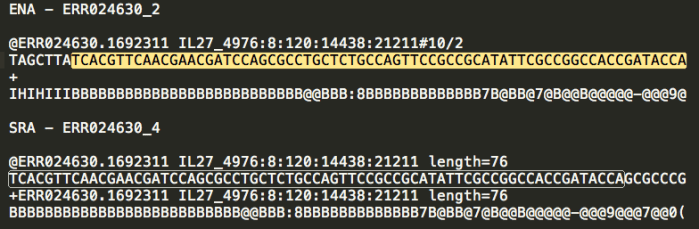
The boxed and highlighted sections are the same sequence.
What on earth is going on? Any ideas?
The data is quite old (2012), so it may be something to do with that? I’m tempted to trust the ENA data, because they got the initial deposition. Also, they return the expected number of fastqs (i.e. 2). But I really like the accession based download syntax of the sra-toolkit so would like to use that if possible. Also, it’s not great if the two databases are giving different results! Hopefully there is an explanation/fix.
Update 2016-01-06
Apparently the data was uploaded in SRF format (which is a new one on me) in some sort of pooled experiment type which is no longer encouraged. After some clues from the very helpful ENA helpdesk, on further investigation the SRA _1 file has illumina barcodes on the 3′ end (and is 82bp), SRA _4 has no barcodes (and is 76 bp). ENA _1 (76 bp) has no barcodes, and ENA _2 (76 bp) has barcodes on the 5′ end! What a mess! The main thing that is bugging me now is why ENA_1 and ENA _2 are the same length, when one of them has the barcode on and the other one doesn’t. So, I think the most trustworthy data will be the SRA data with the barcodes removed? But, what a palava!
Also, here is a script to download data from the ENA ftp site https://gist.github.com/flashton2003/336e67bbc513b0cf3f07





the data at NCBI/SRA was extracted from the SRF as it was described in the Experiment xml which specifies a 1 base technical adapter following a 6 base barcode. ENA helpdesk is spot-on. pooled multiplexed SRF was rarely processed neatly and . and never demultiplexed by the archives (we did demultiplex 454 SFF). based on age and instrument, i’m guessing 2×76 paired end run. not sure what the single base technical read is.
different results are due to the SRF being processed separately at EBI and NCBI. EBI never provided archive format for mirroring (see multiplexed pooled SRF) so NCBI pulled the submitted file and tried loading it using the Experiment spot descriptor. i could reload and force the forward/reverse reads into shape, but i won’t touch demultiplexing.
i fixed this one at NCBI. it will split correctly. you can use fastq-dump -G option to get reads split out by barcode.