We, like many people, are interested in optimal de novo genome assembly. When you assemble a genome you want the best possible representation of the ‘true’ genome. How can we obtain the best possible representation?
In the past we have used VelvetOptimiser to do our assemblies. This assembles with a range of k-mers and returns the best assembly (edit: in terms of N50). This approach produces assemblies with high N50s, which is a handy rule of thumb for assembly quality.
However, like the old adage ‘as soon as a measure becomes a target, it ceases to be a valid measure’, we are wondering whether optimising for N50 in this way provides the ‘best’ assemblies.
So, we decided to assess a couple of different k-mer estimation tools, Kmer Genie and VelvetK, and see how assemblies with their K-mers stack up against VelvetOptimiser. The test sample was paired-end fastq data from a shiga toxin producing E. coli O157 I’m working on, representative of our samples in terms of coverage and quality.
The three different assemblies were then assessed with Quast, a reference (I used E. coli O157 Sakai) based assembly quality assessment tool.
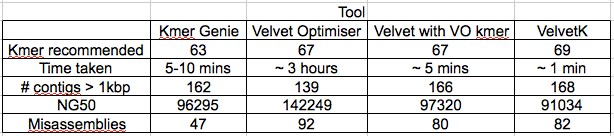
Table 1: Characteristics of the different k-mer estimation tools, calculated by Quast. Velvet Optimiser k-mer range was 21-121, to match the Kmer Genie range. Edit: non-velvet optimiser assemblies were repeated with -exp_cov and -cov_cuttoff set as ‘auto’ with much improved results. Also did Velvet with the velvet optimiser k-mer to compare the improvement of optimiser only.
All k-mer estimation tools gave k-mers between 63-69. As expected velvet optimiser gave a Edit: slightly better NG50.
It is the misassemblies stat that really leaps out. Quast is saying, that relative to Sakai, the VelvetOptimiser assembly had 92 misassemblies, while VelvetK had 44 and Kmer Genie had 12.
So, are there genuine biological differences between the Sakai genome and the sample genome (caused by e.g. recombination or rearrangement), or, is VelvetOpt forcing misassemblies in the quest for a better N50? How to tell? Well, I think it is very difficult (impossible?) to 100% determine this using a bioinformatic approach but we can get a better feel for the answer if we use the paired end information in our dataset.
Our hypothesis is that if a contig is correctly assembled, it will contain a high proportion of properly paired reads. Velvet should take this info into account when assembling – but does it? We mapped the original reads back to the three assemblies using bwa and I wrote a script (using pysam) to calculate the percentage of reads in each 100 bp window (iterating by a single base each time) that are properly paired and produce a plot of percentage mapped along the contig length.
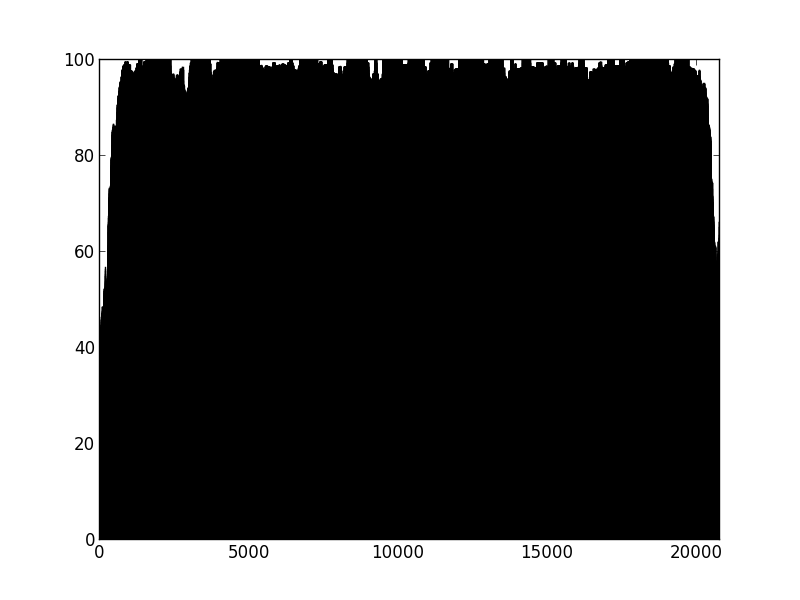
Figure 1: Percentage of properly paired reads in a 10 bp window along the length of the contig. This contig was not highlighted as misassembled by Quast (i.e. was a positive control). X axis is base-pairs along the contig , y axis is percentage of reads properly paired. There are bound to be dips at either end of the contig, length of this dip will be a function of length of DNA fragment sequenced (~700bp in this case).

Figure 2: Percentage of properly paired reads – two unrelated contigs, artificially stuck together by myself (negative control). The junction is at about 4500 bp
So, how does this work on contigs labelled as misassembled by Quast?

Figure 3: Percentage of properly paired reads – contig labelled as misassembled by Quast. There are two dips in the percentage of properly paired reads, one at around 20000 bp, the other a couple of thousand bps downstream.
So, figure 3 certainly looks a lot different from the the figure 2 (negative control), but is also different from figure 1 (positive control). Does this mean that this contig is misassembled? There are a couple of little validations we can do. Firstly, look at this contig in Tablet and there is a series of unpaired reads at the genomic location indicated by the script. Also, where the reads are ‘properly paired’, the two halves of the pair overlap. Secondly, we can look at how this contig assembled in the Kmer Genie assembly – see edit below.
Edit: when exp_cov and cov_cutoff were set to auto in the velvet assembly, the kmer_genie assembly resulted in a single contig which Quast then annotated as misassembled.
This all seems like along way of telling you what you already know – if you want long, questionable contigs then go with VelvetOptimiser, if you want Edit: slightly more conservative contigs, go for Kmer Genie or VelvetK. However, I feel more comfortable with this kind of information rather than just having a black box of assembly.
While chatting about this with Anthony Underwood, he said that he thinks the killer experiment in this area will be an assembly of Illumina paired end reads from a strain for which a Sanger reference is available – then you can call true misassemblies to help calibrate your tools. Either that or a shed-load of PCRs to characterise what it means when properly paired reads drop below a certain percentage.
p.s. the script is very slow (takes about a minute for a 10 kb contig) due to the way I have done the windowing (with a step of 1), there is an option in the comments of the script for changing this but it messes with the display of the graph for a reason I’m not quite sure about yet. The script is very much a work in progress and if I have missed any bugs or made a giant error then please let me know.
p.p.s the script is only in the public folder of my dropbox, as I’m not cool enough for github. yet.





Nice post Phil. An interesting description of the problem and discussion around how to delve deeper. As a side note I’d try the velvet assembly with a kmer of 67 ( as suggested by Optimiser) and check you can reproduce the high N50 score. Is it a product of optimising coverage as velvet does after determining the optimal kmer?
Thanks Anthony!
Yes, good idea, will give it a go.
Ok, so I was doing a disservice to Kmer Genie and VelvetK.
I neglected to set the the cov_cuttoff and exp_cutoff Velvet params to auto (thought would default to auto). Once you include those params, these assemblies get much better and there is less of a differential between Velvet Optimiser and the other tools – updated the table to reflect this.
Pingback: Philip Ashton: Assessment of Assembly - Homologus
As you have reference you can also compare NGA numbers, which will took misassemblies into account. How do the results look like then?
Hi Anton, NGA 50 calculated by Quast:
VelvetK – 11171
Kmer Genie – 11434
Velvet Optimiser – 105873
This is interesting as Quast also says that there is 2 Mbp that is misassembled in the Velvet Optimiser assembly, must be mostly small sections that are misassembled?
Usually the ends of the contigs are of worse quality, yes. However, note that if you have single 2 Mbp contig with single misassembly in the middle, then NGA will be calculated using two contigs of 1 Mbp length. Given that NG50 is already around 100k, it won’t influence the result that much. However, 1.5x drop of NG vs NGA indicates decent amount of misassemblies, yes.
AFAIR, somewhere inside quast’s results directory the extensive contig analysis log is hidden. You might want to study it to check how exactly the contigs are misassembled.
(KmerGenie author here)
Thanks for this blog post, very interesting.
“As expected velvet optimiser gave a much better NG50 – although, as a side note, it is hard to imagine how a difference in the k-mer of 2 from VelvetK resulted in such a dramatic difference in NG50.”
For a given k value, Velvet computes its own estimates of two other values: expected coverage and coverage cut-off.
VelvetOptimizer not only finds the best k but also searches for the optimum expected coverage and coverage cut-off values (maximizing the total basepairs in contigs longer than 1kb), overriding the estimates of Velvet.
This is why VelvetK and KmerGenie assemblies are so different from VelvetOptimizer: VO sets other parameters.
Also, the analysis reminded of the REAPR software (http://www.sanger.ac.uk/resources/software/reapr/).
Hi Rayan,
Yes, it will be interesting as Anthony also mentioned to run velvet with the k suggested by by velvet optimiser and see how much the non-k aspects are contributing to improvement. Will be interesting as most people seem to say that leaving exp_cov and cov_cutoff as auto is the best option.
Pingback: Assembly optimisation – impact of error correction and a new assembler, SPAdes – Bits and Bugs