This is a guest post by Alex Jironkin (@biocomputerist) in the core bioinformatics group at Colindale.
TL;DR: Try assembling your hybrid Illumina-MinION data with smaller number of kmers for better assemblies
By now you probably heard about the new Nanopore technology, it is truly marvellous bit of engineering. The DNA strands are pulled through the pores, changing the current across it, theoretically giving you ability to sequence entire chromosome at once. If you are one of the lucky few that got your hands on MINion to see for yourself if the hype is true, then you are probably wondering what to do with this data. I was lucky enough to get my hands on some Illumina and nanopore data and here is what we did. First, install the nanopore tools and take a look at the data stats:
nanopore stats fast5
- total reads 30327
- total base pairs 117661971
- mean 3879.78
- median 3436
- min 118
- max 26986
- N25 7406
- N50 5433
- N75 3627
Our data is good, lots of reads and mean read length is 3.8kb (amazing 25 times longer than Illumina reads). To get your data in some usable format you need to export it into FastA or FastQ format. Again this is very easy:
nanopore fasta fast5 > all_reads.fasta
or
nanopore fastq fast5 > all_reads.fastq
Take a look at the documentation for nanopore tools, it has plenty of examples.
Next, lets map the reads against one of our existing assemblies (just to get the feel for the data). There are a number of tools that let you map long PacBio/Nanope reads like LAST or BWA. Personally, I like BWA and it’s very simple to use (to map nanopore reads you’ll need the latest version)
bwa index contigs.fasta
bwa mem -x ont2d contigs.fasta all_reads.fasta | samtools view -bS - | samtools sort - mapped_nanopore.sorted
samtools index mapped_nanopore.sorted.bam
First line makes index from your contigs, then we align the reads in fasta format (-x ont2d is important) pipe the output to samtools to convert to BAM and sort the BAM file all in one line. The last command indexes the BAM file. Now you can open it in your favourite viewer like Tablet
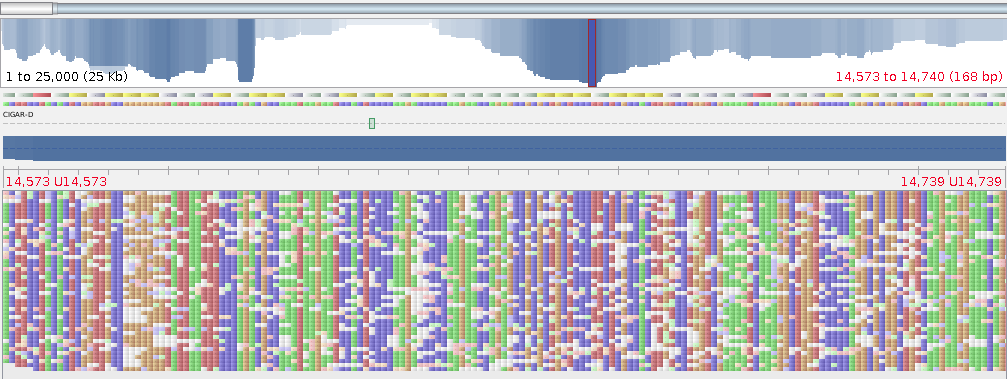
At first glance it doesn’t look great, lots of mismatches everywhere, but you can see features in nanopore reads that correspond to your contigs. It’s ok we are not calling SNPs on this data so it doesn’t matter. We can, however, use the amazing read length to help us resolve the problematic areas that Illumina reads just don’t reach. In other words, we can scaffold our Illumina assemblies with long read length from nanopore. This way you get the confidence of the consensus sequence from short reads and longer contigs. SSPace can scaffold your assemblies, but some assembler will accept long read data to do the scaffolding for you, e.g. Spades (version >3.1.1).
Spades, also uses information from multiple kmer graphs to construct the final assembly. Here is the command I used to assemble my short reads alone:
spades.py -k 21,33,55,63,77,85,87,93,95,97,99 -1 illumina_reads.R1.fastq.gz -2 illumina_reads.R1.fastq.gz -o illumina-11

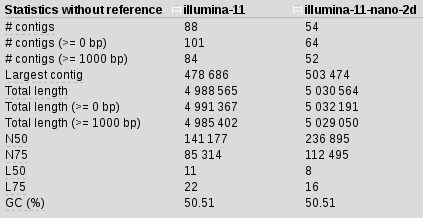
We are using 11 kmers to construct our assembly from illumina_reads R1 and R2. Takes about 5 mins per kmer so 40 mins total (time is not an issue in this case). Using Quast we can compare assemblies and get some stats about them. In this case we get
We are getting good contigs, but this is nothing to write home about. Let’s add nanopore data and see if we can improve this:
spades.py -k 21,33,55,63,77,85,87,93,95,97,99 -1 illumina_reads.R1.fastq.gz -2 illumina_reads.R1.fastq.gz --nanopore 2D_reads.fasta -o illumina-11-nano-2d
We are getting better, but still nothing to be excited about. In fact, I started to get a feeling that something was wrong with the assemblies. I used 11 kmers because longer kmers should improve repeats in the assemblies, but maybe they are somehow interfering instead of helping? I decided to drop the kmer count to 5 instead of 11.
spades.py -k 21,33,55,77,85 -1 illumina_reads.R1.fastq.gz -2 illumina_reads.R1.fastq.gz -o illumina-5
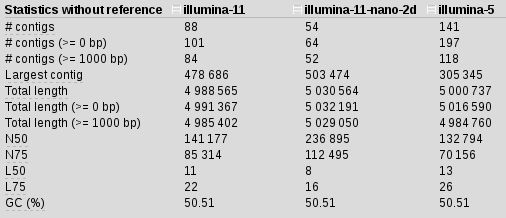
Stats is not great but, what if we include the nanopore data as well:
spades.py -k 21,33,55,77,85 -1 illumina_reads.R1.fastq.gz -2 illumina_reads.R1.fastq.gz --nanopore 2D_reads.fasta -o illumina-5-nano-2d
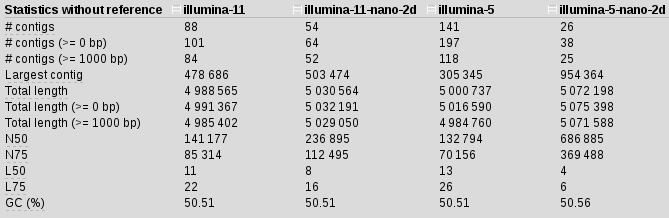
N50 for scaffolds with nanopore have increased by 5 almost 5 times, our contigs are much longer and there are 3 times fewer of them, length of the longest contig doubled.. It seems longer kmers somehow interfering with the assembly and mapping back to the 11 kmer assembly I started to see some weird results, which made me think those started to be misassemblies and not real.
I have tried scaffolding with 1D (forward and reverse treated separately) 2D (two-directional reads) and 3D (two directional, forward and reverse) reads. The summary is here. Our best result was still illumina-5-nanopre-2d.

P.S> There are lots of optimisation that can be done here, but we got what we wanted from this dataset, and so there was no need for us to do them. You can drop kmer to 4 and get 1Mb contigs, and so on.