TL:DR
If you are interested in calling indels with GATK, check out the below. If not, don’t.
——————————————————–
So, this annoying guy asked me to add an analysis of indels* to a paper that has been itching to get off my desk for months. I finally got around to doing this, so thought I would write down the process in case anyone else is interested. I’m going to be using GATK, as this is our standard variant caller and would be simpler to add to routine if we ever want to.
1. Have a sorted bam file.
2. Run GATK (v.2.6.5 in my case), with the following command
java -Xmx4g -jar GenomeAnalysisTK.jar -T UnifiedGenotyper -nt 1 -R reference.fa -I sorted.bam -o indel.vcf -glm INDELS
2a. I investigated the GATK realigner around indels but it didn’t make any significant difference in the one sample I checked.
3. Have a look at the vcf file you just generated. Visually double check the variants it has identified in the BAM using e.g. Tablet.
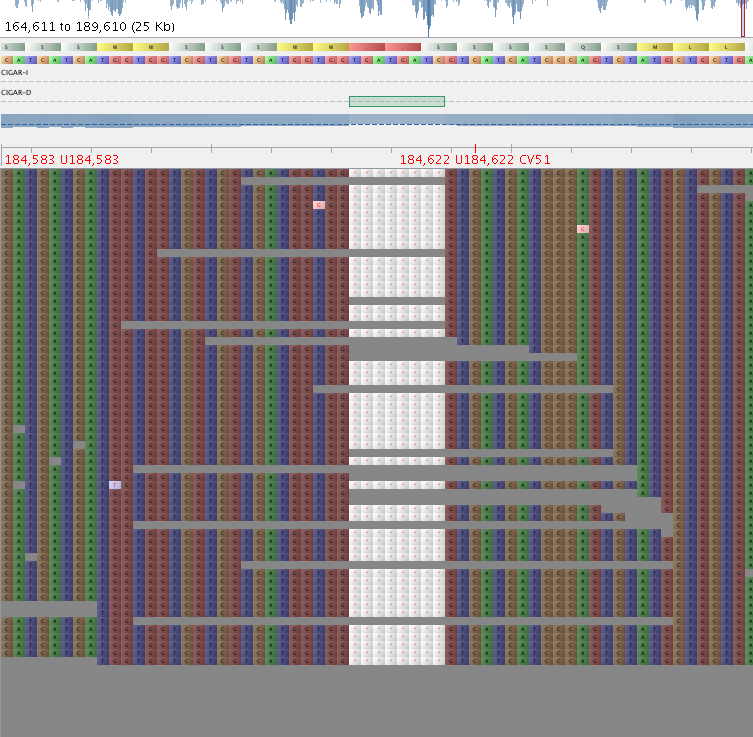
Fig 1: Good indel – QD = 12.43
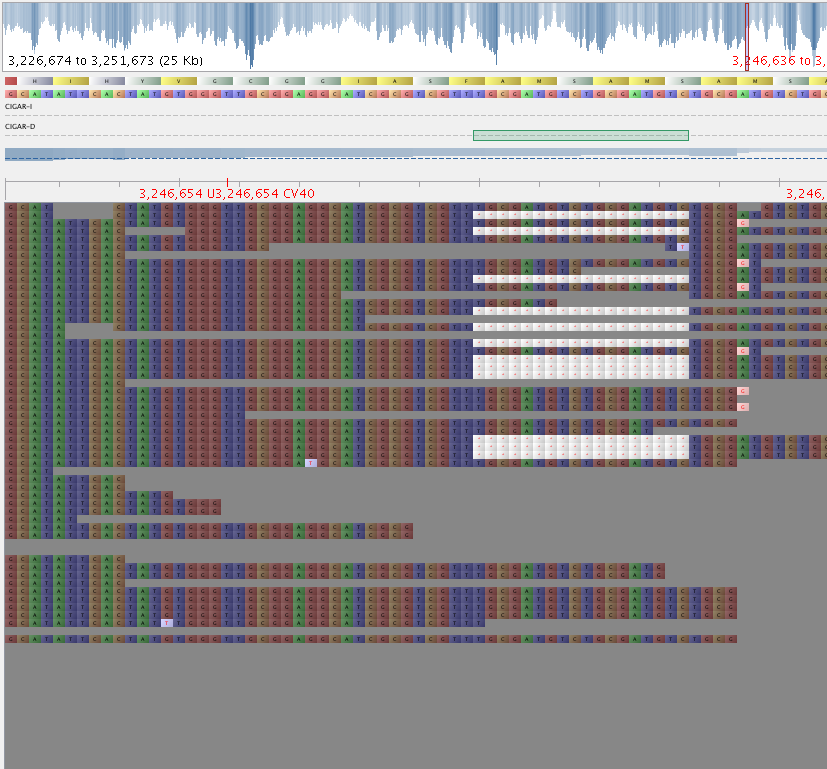
Fig 2: Bad indel – QD = 3.56
4. Parse the indel.vcf and only take variants with a good QD score. I used > 10, as it is a nice round number ;), and variants below this seemed to have some evidence of alternate alleles, while those above did not. DO NOT APPLY YOUR USUAL AD RATIO CUTOFFS, as these will be calculated based on the last position before the indel, and therefore, will not reflect what is going on ‘inside’ your indel.
p.s. if anyone has any more info on gotchas for indel analysis, would be very interested.
* if the indel analysis turns up something really interesting, I may remove this, and claim the idea as my own 😉


Have you tried haplotype caller (now updated for non-diploid organisms) or compared GATK UG to other methods? I’ve been comparing some (freebayes, GATK, varscan, ReadXplorer) and found them to be largely discrepant.
Matt.
Hi Matt, someone in the core bioinfx group did a comparison a year or two ago. What solution have you landed on?
I’ve taken to using haplotype caller at the moment but the comparisons weren’t even consistently discrepant! I noticed an issue with the way most indels are called too in that they’re just concerned with the start position.
I’m trying to write something now for a better comparison of what indels are actually being called! Before, for example, tweaking the k-mer sizes in haplotype caller to refine indel calling.
What kind of discrepancies are you talking about? I always think SNP calling isn’t *that* hard, how can they cock it up?
I know! It was one of those things that during my masters I thought was totally sorted already. There are a few papers that report the concordance of SNPs and indels (sometimes as low as ~20% concordance across numerous methods).
Also there’s an issue with the way matching indels are determined. If you use vcf-compare for example to determine the agreement between two methods it just tells you the intersect of reference positions where the ref or alt is >1, when these indels could be largely different in length to one another. Equally if a method calls a very similar indel but starts a base earlier then it won’t report a match at all. I’ve observed both of these scenarios and am thinking about how to better compare them!
That’s a bit shady. Obviously need to keep track of different indels at the same site.
I guess you have to compare on an indel-to-indel basis and also a disrupted gene-to-disrupted gene basis?
Yes definitely! Working with P. aeruginosa there can be hundreds of indels between strains so I need to write a script to more intelligently work out exactly what various methods are calling!
On a gene basis is something I’ve begun to consider but I’m not going down that rabbit hole just yet!
Hi,
I’m try to use GATK to call genetic variants (SNPs and Indels) in bacterial genomes, but it seems that it is not working for me: using samtools mpileup / bcftools I obtained around 30 SNPs manually checked (including Indels), but with the command line that you suggest and different other GATK tools the maximum that I got is 6 SNPs.
Do you have any idea what could be happening?
Thanks in advance,
Miguel