This is a blog post by Lauren Cowley
———————————————————————————
TL;DR?
- A confusing difference in trimmed/non-trimmed velvet phage assemblies raised suspicions that it was a mixed phage culture
- Spades assembly seemed to show more than one phage present in the culture
- I concluded that spades is better than velvet at showing a representation of the whole sequenced sample but velvet will give you a better assembly of the dominant coverage sample
———————————————————————————
Phage 15 is 1 of a set of 16 phages that are used in the phage typing scheme for VTEC O157. I am trying to sequence the whole set and the majority of them have proved no issue in gaining enough DNA, sequencing and assembling into contextually sensible genomes. Most have assembled into 1 or 2 contigs but Phage 15 was assembling into >50 contigs on the first attempt so we re-sequenced the little tyrant as we assumed there was a contaminant. We were dismayed to find the same issue on the second attempt. It is known that Phage 15 should be very similar to Phage 1 in genomic content and the original assembly on the second round of sequencing showed a very high rate of sequence similarity between them but was still in >50 contigs. This was done before trimming and using velvet optimiser. The below image shows a mauve alignment of the Phage 15 assembly pre-trimming compared with Phage 1.
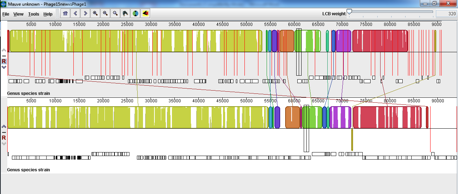
I then proceeded to trim to try and improve the n50 with trimmomatic using minlen and trailing parameters and got this assembly with the trimmed version on top and non-trimmed below;
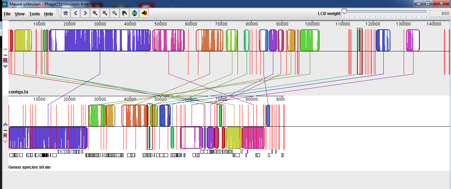
This confused me no end as I had improved the n50 but seemed to have gained an extra 50kb which in a phage expected to be 90kb is a significant increase! We started to think that we might have a mixed sample of two very similar phages and that the second low frequency phage would be overlooked in untrimmed data but would be incorporated into the debruijn graph if the data had been trimmed.
My supervisor suggested we try SPaDES as it is thought to have a better low frequency k-mer elimination step. We also wanted to just take a subsample of the reads as there was extremely high coverage so subsampled to x150 coverage. We got a much more satisfactory outcome, the image below shows Phage 1 on top and the new SPaDES assembly of Phage 15 below;
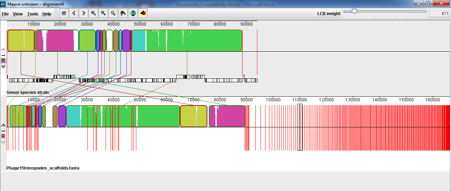
We did a blast all vs all of the contigs in the new spades assembly and found that the sequences in the small contigs found at the end of assembly were almost all in the larger contigs that were similar to Phage 1 found at the beginning of the assembly with just a few regular SNPs between them (Mauve struggles to show paralogous sequences). We think that this has confirmed our original theory, that it was a sample of two very similar phages with the typing phage present at higher coverage that could assemble but without a good low frequency k-mer elimination step these two phages were assembled into erroneous contigs together that added an extra 50kb to the assembly.
It is puzzling that this has not happened before the sample was trimmed but it is probable that only the top of the coverage bell curve was taken into account when assembling that dataset which would have consisted of only the true phage reads that were at high coverage. The elimination of reads during the trimmomatic step would have incorporated more of the low coverage reads into the top of the bell curve after the redistribution and these would have been incorporated into the assembly.
SPaDES has proven an advantage over velvet in its low frequency k-mer elimination and has given us a much clearer solution to the problem. From similar experiences with other phages it appears that Velvet is good at assembling the most dominant in the sample but SPaDES gives you a broader picture of all the other things that you have sequenced in your sample. For example, I had previously sequenced and assmbled with velvet Phage 1 of the typing phage set and it had assembled into 2 neat contigs that matched what was expected for that particular typing phage. I decided to reassemble that phage with SPaDES after what I had experienced with Phage 15 and was intrigued to see a massive difference, see below (SPaDES assembly above and velvet assembly below);
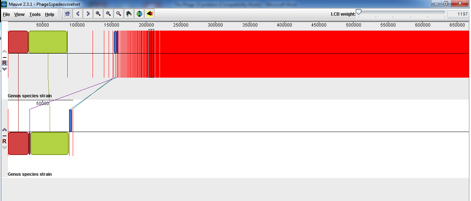
At first this scared me shitless but after looking a little deeper into what was in the other contigs and the horrendous red smear it seemed that the VTEC O157 propagating strain that we use for propagating the typing phage had also released the Lambdoid shiga toxin phage and a few cells of the propagating strain seemed to be included in the red smear. This was evident from the Mauve alignment I did of the SPaDES assembly (above) to the propagating strain (below).
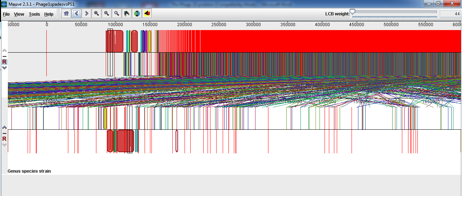
This shows that the typing phage is not present in the genome of the propagating strain but the shiga toxin phage has been induced from the propogating strain and been included in the sequencing. Clearly during the propogating step the shiga toxin phage was induced and included in the typing phage 1 sample and dna extraction for sequencing.
This makes me think that I could start to look at Velvet and SPaDES as two similar but slightly different tools where velvet can be used to give you a good assembly from the highest coverage genome but SPaDES can be used to give you an assembly of all that can be found in the sample.





“At first this scared me shitless” – Lauren, please come and run my lab.
lol isn’t it in chicago? I’d get too fat on chicago pizza 🙂
We’d get fat together. It will be emotional.