As the final part of my assembly optimisation I was interested in the impact of Titus Brown’s Khmer/Diginorm approach on speed and quality.
Ten samples (VTECs sequenced on GAII) were assembled with a variety of pipelines:
- SPAdes corrected fastqs, assembled with SPAdes
- Khmer-ed fastqs, SPAdes corrected, assembled with SPAdes
- SPAdes corrected fastqs, assembled with Velvet
- Khmer-ed fastqs, assembled with Velvet
So, the two things I’m testing here are:
- Whether Khmer/Diginorm can speed up SPAdes assembly without significantly impacting the assembly quality
- Whether Khmer could be used in front of Velvet as an alternative to the much slower SPAdes correction step.
I used a two step Khmer approach, normalize-by-median to coverage 20 with a k-mer of 20 followed by filter-abund (for more details see the Khmer package). After Velvet assembly the contigs were corrected with REAPR to obtain corrected N50s (cN50). SPAdes was used without the –careful flag and with four k-mers (21, 33, 55, 77). No REAPR correction was done on these as in previous work there no significant misassemblies with SPAdes.
Results!

Table 1: Impact of Khmer reduction on N50 and time to assemble of SPAdes
Khmer reduction of reads certainly speeds up assembly, reducing the time taken from 40 to 15 minutes, if you are assembling hundreds of genomes then this is a tasty decrease (need to add a few minutes to this time for the Khmer steps, these are very quick though). However, it also reduces the N50 by 30%. If I had to take a quick guess as to why, I would say that either SPAdes requires high depth or perhaps that Khmer is not doing enough to take the paired reads into account.
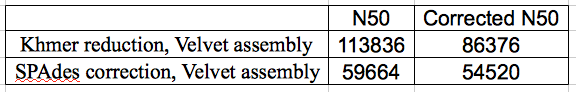
Table 2: Comparison of Khmer and SPAdes correction for N50 and corrected (by REAPR) N50.
With the Velvet assembly, Khmer reduction improved assembly (corrected N50) significantly. Previous work has shown that SPAdes correction improves Velvet assembly cN50 ~3 fold (Musket correction also works well in this regard). Khmer reduction of the reads increases that another 58%. However, if you look at the raw N50 compared with the REAPR cN50 you can see that Velvet is making a lot of spurious assemblies compared with the SPAdes-Velvet approach and that you would really want to check your contigs after the Khmer-Velvet approach.
On balance, I think that the SPAdes correction and assembly is the best option, although I would be interested to see the impact of different Khmer coverage cutoffs on these stats (higher coverage might help keep the SPAdes N50 up?).





The Khmer reduction, Velvet assembly N50 of 113836 is remarkably close to the N50 of 112343 with SPAdes correction and assembly. Admittedly after correction with REAPR the corrected N50 is 86376 with Velvet. Can we be sure that REAPR wouldn’t have corrected the SPAdes N50 and can we even be sure that REAPR is reporting correctly? Is it worth aligning the contigs with Mauve to a reference and seeing where the contig breaks are?
Also with Khmer approach, could you increase the normalize-by-median to coverage to 40/50 and see if it improves things?
In the previous post REAPR correction didn’t change a single N50, so I left it out here. Could definitely do with some validation of REAPR, what is Rediat up to? 😉
Yes, that is the next obvious experiment, this was just a first glance. Need to get on with other stuff though.
Hi Phil, thanks for posting! I am interested in what Anthony asked too 🙂
Right, diginorm has unknown impact on scaffolding, which is just a fancy way of saying that scaffolders make a number of assumptions about coverage that diginorm trashes. Something we’ll look at down the road.
Also, how did you choose the k? Diginorm changes the k assumptions.
We’ve got one euk genome paper using diginorm in 2nd review, and several indications that diginorm can prove genome contig assembly by increasing sensitivity. So I don’t think genome assembly is a lost hope, but I believe that genome assemblers are more highly tuned than transcriptome and metagenome assemblers and so may break under the coverage changes imposed by diginorm.
Thanks Titus!
For the Velvet assemblies I chose k using velvetk on the shuffled, Khmered, paired fastqs (removing the orphan reads might crocked the assembly). For SPAdes I used the same defaults as I usually do – 21, 33, 55 and 77.
I would definitely be interested in a more thorough examination of Diginorm for genome assembly, ideally we want something quicker than SPAdes (or SPAdes, but quicker).
I asked Philip to provide SPAdes logs from the both cases and here are the observations which can be deduced from the logs:
1. Note that the maximum read length here is just 133. So, k =
77 may be a way high. Probably 67 should be used instead.
2. khmer dataset definitely contains more places, when there are
significant coverage drops (e.g. on full dataset SPAdes closed 27 gaps
and on khmer-reduced 170). By “significant coverage drop” I mean the
place when only 2 reads overlap by = 21).
3. khmer-reduced dataset has only 3x average k-mer coverage at k = 77,
which is really small… Due to this SPAdes repeat resolver might not
have enough pair information to resolve the repeats
So, basically, with khmer here one’s playing on the “boarder of the coverage” and random fluctuations might significantly influence the results…
Also, N50’s are shown – but are the overall contig lengths same?