We have seen significant differences between assemblies of shiga toxigenic E. coli genomes where the DNA was extracted with different protocols. In those experiments, I used Velvet for assembly and I wanted to check whether the difference we were seeing between the different extraction methods was somehow a Velvet artefact.
The recent GAGE-B paper showed that the SPAdes assembler gave the best results at 100x coverage (N50 > 2x that of Velvet). SPAdes is a relatively new assembler, originally intended for use with single cell sequencing data that also performs well with standard data. It takes an interesting approach involving the use of paired de Bruijn graphs which allow the integration of read-pair info at a much earlier stage of assembly than most de Bruijn graph assemblers. SPAdes recommends error correction prior to assembly, BayesHammer, from the same group in St Petersburg, is bundled with SPAdes. Quake is also recommended as an alternative. As an aside, SPAdes was very straightforward to install and run.
In addition to N50, assembly quality was assessed using REAPR, a reference free tool from the Sanger. We had some teething issues getting this set up but the developer, Martin Hunt, was helpful. REAPR assesses an assembly by mapping the reads to the genome and looking at whether the paired information is congruent with the assembly. It then breaks the assembly where it detects errors and returns a corrected N50. For a few samples I compared the number of misassemblies called with Quast, a reference based assembly assessment tool we have used in the past and REAPR.
So, what were the results?
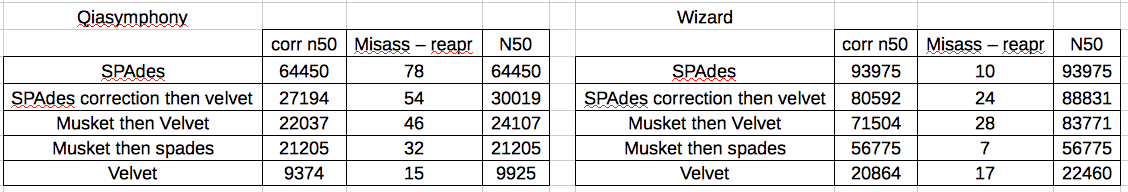
There was a difference between how the Qiasymphony data and the Wizard data were handled by the different approaches.
For the Wizard data, SPAdes gave the best results (corrected N50), nearly 5x better than Velvet. However, this isn’t really a fair comparison as SPAdes has an error correction step. Therefore I used Velvet to assemble the SPAdes corrected reads and obtained an N50 roughly equivalent with the SPAdes.
Then, to see how BayesHammer compared with other error correction tools I corrected the fastqs with Musket and assembled with Velvet and SPAdes (without error correction). These assemblies varied but were all much better than pure Velvet, with Musket -Velvet 3rd, followed by Musket-SPAdes.
The take home message is – error correct your reads before assembly!
For the Qiasymphony data, the approach that gave by far the best results was SPAdes, with SPAdes-Velvet, Musk-Velv and Musk-SPA all significantly worse and much of a muchness. Every method including error correction significantly improved on pure Velvet. To me, this indicates that the problem with the Qiasymphony data is a combination of something amiss with the insert size distribution and an error profile that is corrected by BayesHammer but not Musket.
In terms of misassemblies – REAPR called an average of 9 for SPAdes and 23 for Velvet. The misassemblies called by REAPR didn’t impact the N50 (or N90) of any of the SPAdes assembled genomes, indicating that these misassemblies were all in very small contigs. For the Velvet genomes, the REAPR correction decreased the N50 in 20 of 24 by an average of 7%, indicating that REAPR is breaking larger contigs in Velvet assemblies.
In terms of reference vs reference free assembly assessment – there was a correlation (R-squared) of 0.15 between Quast and REAPR in terms of total number of misassemblies called (n = 20). Not very good! I definitely prefer the REAPR approach, I’m somewhat wary of misassemblies called in comparison with a reference.
I should mention that SPAdes takes longer than Velvet, error correction takes around an hour and each k-mer takes 5-15 minutes (depends on file size). However, Musket is also quite slow, so if you are including an error correction step in the Velvet assembly it is roughly equivalent. I’m going to look into the impact of khmer/Diginorm on SPAdes assembly – there will definitely be a speed improvement, so will be interesting to see what happens to cN50.
TL:DR? Redux: try SPAdes for genome assembly. Correct your reads before assembling.





Very informative post. Did you run velvet with -cov_cutoff auto?
Thanks Rayan!
Yes I did. These were particularly bad Velvet assemblies so I wouldn’t expect that kind of improvement as standard. They were chosen as I needed pairs of genetically similar strains that had been extracted with the Qiasymphony and Wizard kits.
Great post!
Have you tried stand alone read error correction from ALLPATHS-LG?
http://www.broadinstitute.org/software/allpaths-lg/blog/?p=559
Thanks!
No I haven’t, would you recommend it?
Yes, it is easy to run and highly recommended by Dr. Rayan.
Click to access 2013-evomics-assembly.pdf
My last name is Chikhi but I really appreciate the referral 🙂
Note that I am not alone saying this.. the GAGE paper also stated it: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3290791/
Pingback: Impact of Khmer/Diginorm on SPAdes and Velvet assembly – Bits and Bugs
Been playing around with this. Have you noticed N’s in your contigs? (I checked the input sequences)?
Have you noticed N’s in your contigs? This is strange (I have checked the input pairs)…
Hi Mitchell, yes I did see some Ns. From my recollection of what the developers said that is where there is ambiguities in the assembly, for example paralogous genes. Using the ‘careful’ option or version 2.5 might reduce the number of Ns.
How did you find it in terms of N50?
Great post! Let me know if/when you get diginorm results — I still want to know even if they’re poor 🙂
Hi Titus, thanks!
I had a quick look in the next blog post, still lots of experiments I would like to do…
https://bitsandbugs.org/2013/07/02/impact-of-khmerdiginorm-on-spades-and-velvet-assembly/