As our project ramped up, we moved from a manual extraction method (Wizard) to a high throughput method (Qiasymphony). We thought that it would be prudent to check that the sequence data obtained by the two methods was equivalent. Therefore, on a GAII run of 96 strains we sequenced 72 isolates that had been extracted by Wizard and 24 isolates that had been extracted by Qiasymphony.
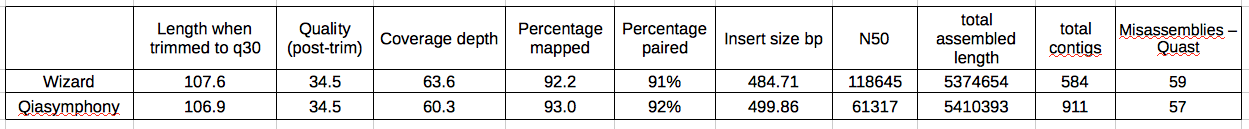
Table 1: Quality, mapping and assembly statistics of Wizard and Qiasymphony extracted DNA
DNA extracted by Wiz and Qia give reads that are equivalent in length and quality. When they were mapped to a reference genome (Sakai) with BWA they gave equivalent coverage of the reference, equivalent percentage mapped, percentage properly paired and average insert size between those pairs.
The only way in which data resulting from DNA extracted by Wiz and Qia was different was in the quality of the assemblies. When I assembled these samples (velvetk -> velvet) there was a significant difference in N50, with the Wiz DNA giving an N50 of 118645 and the Qia DNA giving an N50 of 61317. The Quast results imply that this difference probably isn’t due to a larger number of erroneous assemblies in the Wizard samples.
It also can’t be explained by strain to strain differences as the difference was maintained between strains that were 0 SNPs apart and where the same strain had been sequenced more than once. Also, the difference was maintained within lanes so it wasn’t per-lane variation.
To really get to the bottom of this, I need to look at the contig differences between the assemblies – where is the Wizard assembly contiguous while the Qiasymphony assembly is broken.
I have also looked at the impact of an alternative assembler on this phenomenon, but that is a different post.

Pingback: Assembly optimisation – impact of error correction and a new assembler, SPAdes – Bits and Bugs