This is a guest post from Anthony Underwood.
——————–
I’ve been exploring the use of IQ-TREE for rapid ML tree calculation. I’ve had reservations for a while when using RAxML since even with multi-threading it can take hours to run and have often instead submitted them to the CIPRES supercomputing facilities at phylo.org where the results are usually returned in under an hour. This again makes me uncomfortable since it is an external service and is not necessarily something we can rely on.
Some of the features of IQ-Tree are
1) A novel fast and effective stochastic algorithm to estimate maximum likelihood trees
2) An ultrafast bootstrap approximation to assess branch supports which is 10 to 40 times faster than RAxML rapid bootstrap and obtains less biased support values.
3) An ultrafast and automatic model selection which is 10 to 100 times faster than jModelTest and ProtTest.
It has a FAQ which describes how it treats ambiguous characters (similar to how RAxML does) and how to interpret the bootstrap support values – they suggest only trusting clades with >= 95% support. In terms of compilation, it requires a recent version of CMake (2.8+) and GCC (4.6+), apart from that compilation is fairly straight forward.
This command line option (once IQ-Tree is installed) will test which evolutionary model is best, construct an ML tree using this model and perform 1000 bootstraps. For an alignment with just SNP positions this will in my experience take minutes and for an alignment containing 100 average bacterial full genome length sequences , a couple of hours.
`iqtree-omp -s alignment.fas -nt 4 -m TEST -bb 1000`
The results look promising with tree topology, branch length, and support values showing broad correlation (see below for data). The fact that this is an actively developed piece of software (https://github.com/Cibiv/IQ-TREE), with good documentation and some good peer reviewed papers (http://dx.doi.org/10.1093/molbev/msu300 and http://dx.doi.org/10.1093/molbev/mst024) gives me confidence to try this for the next few phylogenetic analyses I need to run
.

Neisseria Tanglegram (RAxML on left, IQ_TREE on right)
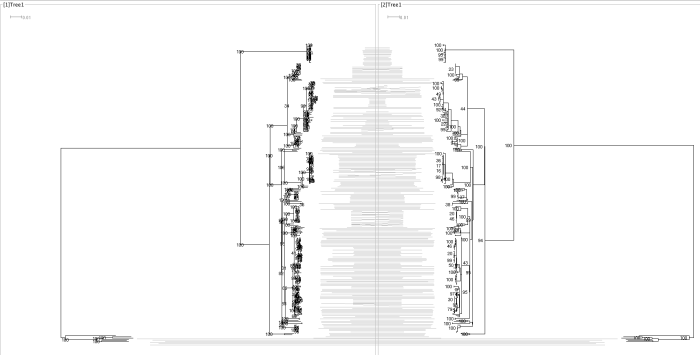
Shigella sonnei tree((RAxML on left, IQ_TREE on right. Thanks to Tim for the data)





Interesting post! I’d also add some thoughts on why I think Iqtree may be better regarding some flexibility on some parameters, IMHO (I’ve nothing to do with its software developers, probably never met them personally either):
– RaxML uses fixed branch lengths across partitions (not 100% sure RaxML doesn’t have this option now, a while ago it didn’t), Iqtree allows completely unlinked, or proportional, or unfixed model. The proportional model is more recommended than equal branch lengths across partitions (ratios among brlens in a partition are kept the same in another except for a constant of proportionality), because it accommodates more properly partitions having different rates.
– UFBoot support in Iqtree has a much better correlation with probability of clade being correct than regular bootstrap (but not known compared to previously known support measures).
– Iqtree permits using (by the way, even testing) molecular models, while RaxML is stright GTR (plus with or w/o rate heterogeneity).
– Iqtree has better flexibility at defining mixture models (can be important for minimizing compositional biases).
– Iqtree permits the use of heterotachy models. It’s probably the 1st software to allow for different lineages having different rates across partitions that is fast for large phylogenomic datasets.
I may be forgetting something… in any case, it’s definitely worth testing.
Hi Jose, thanks for the detailed comment!!
A useful post!
However , I’m wondering what tool you use to compare the two trees generated by RAxML and IQ_TREE and present the comparison result in a face-to-face style? Thank you!
Hi Duo Xie, I imagine Dendroscope was used as per this blog post
https://bitsandbugs.org/2013/11/20/how-to-do-a-dendroscope-tanglegram/
Dear flashton,
I have read this blog and I do it as you posted in Dendroscope ,and it works very well!Thank you very much !!
Woo hoo! Happy to help 🙂