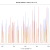Credit for this post is really due to Peter van Heusden, Torsten Seemann, (and Anders Gonvalces Da Silva has had lots of useful discussions with Torsten about this issue), Conor Meehan and Leonardo de Oliveira Martins.
TL:DR
- Two ways to easily give IQ-TREE the number of constant sites:
- From Leo – For whole genome phylogenetic trees, you can feed IQ-TREE your whole genome alignment (including constant sites) and it will run in a reasonable time if you pass it the following params `-t PARS -ninit 2`. If you use the IQ-TREE default parsimony options then it doesn’t remove repeated patterns (including the many invariant positions), and will take a very, very long time for larger alignments.
- From Peter/Torsten – Feed IQ-TREE the output of snp-sites -c as the sequence, as well as the number of invariant positions (output by snp-sites -C, new feature added by Peter) via the -fconst feature `iqtree -fconst $(snp-sites -C genome.aln) -s <(snp-sites -c genome.aln)`
Background
Bacterial whole genome phylogenetics often involves generating a consensus genome from your data + an appropriate reference genome, then combining these consensus genomes for different isolates, extracting the variant positions and using them to build a phylogenetic tree.
One problem with this is that the whole genome alignment usually consists primarily of invariant (monomorphic) sites. If you don’t take this into account when generating your phylogeny, the branch lengths of your tree will not be correct, and the topology of parts of the tree with short branches will potentially be affected.
The most common solution for this is to use ascertainment bias correction. However, there has been some rumblings for a while now that this is not an appropriate method. It should be self-evident that if we throw away information, one should not expect it to be magically recovered. I was recently privy to an interesting discussion of this issue, and thought it would be good to share the outcome of the discussion.
A version of the full discussion, which has been edited for length/relevance can be seen below. This contains further technical details which may be of interest to some.
First question from Peter:
“basic question for you all – when doing SNV based trees, I apply the SNV to the reference I mapped to and then make a MSA by concatenating the reference and the newly generated ref + SNPs genomes (this is assuming 1 FASTA record for reference, 1 FASTA record output per sample). then I pass this MSA to iqtree, using the sample name as the FASTA ID. I’ve got my own script for this but am now proposing to use bcftools consensus for this step (after I filter out non-SNV from the VCF). is this the “standard procedure”? btw I’m working with M. tuberculosis so things are somewhat simpler than I think what others face…:”
Torst:
I count the invariant (monomorphic) sites and use -fconst a,c,g,t
Conor:
“You do need to do asc bias correction for sure and it can affect topology if branch lengths are short.”
“Substitution models are primarily used to deal with multiple mutations at a site and scale branch lengths accordingly, so you end up within estimated substitutions per site for ML trees.
If you use a model that incorporates base frequencies (e.g. HKY or GTR) then the rate matrix will be heavily influenced by the % of each base in the alignment. SNP only alignments have wrong base frequencies, affecting short branches where only 1 or 2 SNPs define them.”
Torst:
“Fconst is really just a linear scaling of the branch lengths.”
Leo:
“One thing to consider is that a column composed of “AAAA” is distinct from one composed of “AANN” or “AA-A”, although all 3 of them will be removed by snp-sites -c, and I think all 3 will be reported as constant by IQTREE. They are distinct in that they represent 3 “site patterns” and add therefore to the likelihood calculation. Identical site patterns (constant or not) should not affect the timing too much. Unless you use partitioned/spatial models.
IOW there is a grey area between variable sites and constant sites, which are the sites with ambiguous states (that are, as you have seen, important in estimating the transition matrix and thus branch lengths).”
Peter:
“ok IQ-TREE with 69 Mtb sequences, variable sites only…. 891 sites… 1696.4 seconds . Same with complete 4.4 MB genomes, 4905.923 seconds – so ok it is not entirely removing the invariant sites from the calculation. Of course the quality of the tree (number of warnings about near-zero internal branches) is much better with the latter…”
Conor:
“Ok I looked into the difference between ASC and Fconst in IQ-TREE and indeed it is as I suspected. ASC is doing a Lewis correction to the likelihood score; this means it just modifies the likelihood calculations to be a conditional on the site being variable or constant. It doesn’t know if it is an A or C or G or T just a blanket correction. Fconst is doing a per site addition of constant sites which are then fed into the rate matrix and frequency calculations. In RAxML these are both put down as ascertainment bias correction whereas IQ-TREE separates them
So for SNP data I would recommend -fconst and not ASC, reserving that for morphological data”
Leo:
“Practically all implementations of the likelihood function (all “professional ones”, at least ) use the site pattern weighting since it’s natural to calculate the log likelihood as f1 LnL1 x f2 LnL2 (where f are the frequencies of patterns and LnL is the partial log likelihood of a pattern at a fictional root). The paper above describes a way to applying the same thing to internal nodes. iqtree uses the pll library, which may have incorporated the internal node repeat algo?
The speed decrease when going from variant alignment (VA) to full alignment (FA) is most likely because
FA = VA + 4 + NA != VA + 4
where NA is the number of site patterns including ambiguous characters (N, –, and friends). I.e. the full alignment does not contain only four extra patterns, but a lot more if you consider a “fifth state” besides ACGT. The likelihood of “AAAA” is different than for “AAA-” and also distinct from “AA–“. To confirm this I would ask @Peter van Heusden to see if these characters are present in FA and in VA.
Other factors include the reading overhead (to create the site patterns based on the big alignment), the correction overhead (not sure about this), and maybe the scaling factor (to avoid underflow, sometimes the LnL is rescaled, and larger alignments have smaller LnL; but this should be over nodes, not sure if applicable).
I just tested on a small data set and concatenating the alignment with itself several times did not change the speed in more than ~5%. Therefore when going from ‘full’ to ‘variant’ you may be losing some information (indels, ambiguity) that cannot be replaced by ascertainment bias correction, from what I understand.”
Conor:
“I go back and forth between running full alignments vs SNV+ASC. Do you think the increase in overheads with the full alignment is always justified?”
Leonardo de Oliveira Martins:
no, I personally avoid whenever possible to use SNVs only (with or without ASC) b/c I have trouble interpreting the branch lengths. But I see a few cases where one might prefer it:
- As mentioned, for the time scales involved you may assume that events are rare (no back-mutations etc) and therefore it’s enough to look at the SNVs. (Although in this case it’s hard to prefer likelihood over parsimony, distance)
- life is short. And from what I see in the simulations inspired by @Peter van Heusden comment, something is not working as expected for very long alignments. (I’ll ask around about this)
- the topology will be at least close to the best one can get. Therefore one could start with a SNV+ASC to get a good starting point for a full alignment later on?
- some other case I haven’t contemplated given my limited experience with bacterial strain-level analysis…
I still see, however, room for improvement in handling ambiguity — both in the methods, that treat equally “-” and “N” with few exceptions, and in the analyses, since in theory they help estimating the rate matrices but are not contemplated in an SNV alignment.
Conor:
“I think handling ambiguity correctly is ‘the next step’ for phylogenetics. Many/most programs just treat them as gaps which is not the same thing. I am in full agreement on your points about using SNV. I think like going from genes to genomes wide SNPs, once the methods get even more efficient, we will be going to ful genomes only and leaving this SNP+ASC behind.”
Torsten:
Thanks for the PR @Peter van Heusden
Now one can do this from a snippy-core result:
iqtree -fconst $(snp-sites -C core.full.aln) -s <(snp-sites -c core.full.aln)
Docs here
Leo:
To give a happy ending to this story, I found out why iq-tree running time was linear with the sequence length (it should be sub-linear, or in my case, constant): it’s the initial parsimony trees estimation. Apparently their default parsimony calculation (which relies on the PLL http://www.libpll.org library) doesn’t take advantage of site patterns, having execution time linear w/ the sequence length.
IQ-TREE finds parsimonious tree twice, by default: 1. to create an initial tree, and 2. to populate an initial tree set. (1) can be modified with “-t” and (2) with “-ninit 2”.
Fortunately the authors recently added an option “-t PARS” to use their own algorithm for initial parsimony tree, much faster (not documented?). Other options for estimating the initial tree are “-t BIONJ”, “-t RANDOM”, or “-fast” (which, however, uses the PLL version once). So I got the expected results with “-t PARS -ninit 2”, for instance.
Tests – I ran a small set of IQ-TREE tests on 40 Lineage 4 TB genomes. Full is the whole genome alignment, “Leo” is with the `-t PARS -ninit 2` params passed to IQ-TREE, snp-sites is where the tree was fed the output of `snp-sites -c`.
Full – 2m20s
Full Leo – 1m20s
Snp-sites – 58s
Snp-sites leo – 48s