TL:DR?
- Lighter is an excellent sequencing read error correction tool, fast (90 seconds for 700 mb unzippped fastq) and well engineered (install was completely painless)
- It significantly speeds up assembly – 20% in my quick benchmark using a Salmonella genome
- It reduces the number of positions that have an AD ratio of <0.9, remember – every SNP is sacred!
One of the great things about bioinformatics is that a single new tool can open up a world of possibilities. Lighter, a read correction tool from Li Song et al (from the lab of Ben ‘bowtie’ Langmead), could be one such tool. I hope they wouldn’t mind me saying that the strategy is not novel. You can remove errors from your reads by discarding/altering reads that have a low frequency relative to other similar k-mers i.e. if you have 50 occurrences of ACTGACTAGCTAGCATCGAT and 1 occurrence of CCTGACTAGCTAGCATCGAT, then you can make the supposition that the low abundance k-mer is the result of an error, and discard/amend it. Even if the general idea is not new, this implementation could be a boon for people who want to do genomics with an emphasis on speed of analysis.
However, it is a bit scary to start messing about with your reads! You are changing it to ‘what you think it should be’, doesn’t sound very scientific! However, sequencing has errors, and why would you not squash these if you can? Obviously taking care to ensure that nothing untoward is occurring. There are also numerous other advantages that come from error correction.
- Better assemblies – fewer errors means fewer kmers, means a simpler de bruijn graph. This should lead to quicker, better assemblies. One of the Unique Selling Points of Lighter is that it is FAST. Previous read correctors I have tried (Musket, Quake, Hammer (from spades)) were slooooooowwww). The assembly speed up from error correction was not sufficient to make up for slow error correction. Lighter is quick enough that it might provide a net gain in time, while bringing the advantages of error correction.
- Better mapping – this is the primary reason I’m interested in Lighter right now. We have a problem with some mixed Salmonella isolates being sequenced. These mixed Salmonella have lots of mixed positions which lead to those positions being ignored when it comes to gathering high quality SNP positions. If you ignore it in one strain, you have to ignore it in all strains, which can collapse your tree. A while ago Tim suggested that error correction (which really just removes minority alleles) could help with this problem.
- Better compression? Reference based compression like CRAM stores reads based on their similarity to a reference genome. The fewer errors in the reads, the less ‘information’ there is to store, the smaller your CRAM files (and BAM files?) will be.
So, I have had a quick look today at Lighter and it’s impact on mapping and assembly.
Firstly, mapping.
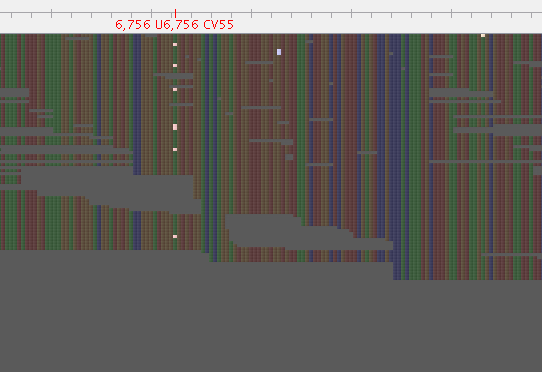
Figure 1: Before Lighter
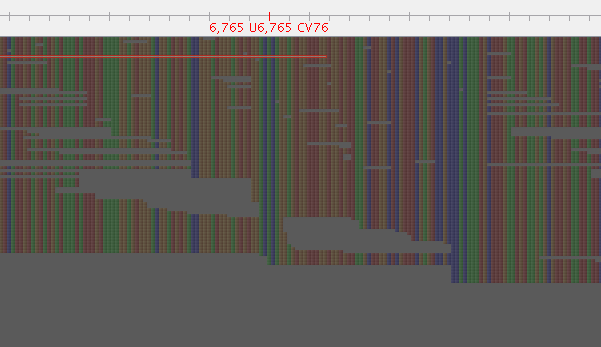
Figure 2: After Lighter
I used Lighter with an alpha of 0.05 (lower than the 0.1 recommended by their rule of thumb) to obtain this result, an alpha of 0.1 seemed to leave more of these mixed positions.
So, that is a snap shot. When we parse the VCF looking for heterozygous calls (0/1), there is a decent reduction, with 1090 in the VCF derived from un-corrected reads and 456 in the VCF derived from corrected reads. This approach is worthy of further examination for our mixed sample problem.
Secondly, assembly.
I had a very quick look at assembly of corrected and non-corrected reads from a Salmonella genome. Spades params were a single k-mer (55), no error correction, not careful, single thread.
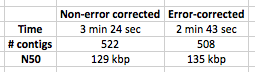

Table 1: Speed and quality of assembly with corrected and non-corrected reads. EDIT – I have added in the stats for assembly for correction and non-correction on untrimmed reads i.e. no quality trim.
I have to say, I’m very happy with these results. 20% speed up in assembly with a 90 second error correction step, with modest improvements in N50 length and number of contigs as well! The amount of time saved in the assembly will increase as multiple k-mers are used to assemble.
Edit – wow, I would have bet at least £10 that untrimmed reads would give a worse assembly, but at least in terms of N50, the N50 of untrimmed reads is better, and the error correction makes it even better! Does take longer though.
Finally, compression.
The corrected bam was actually 2 Mb larger than the uncorrected bam. Not sure why…
In summary
Lighter looks really interesting but I want to be a bit more certain that nothing is being lost before using it in anger.