TL;DR
- Pseudofinder run using the bakta database has a good sensitivity and decent postive predictive value for detecting pseudogenes.
- Pseudofinder run with the bakta database can differentiate between gastrointestinal and extraintestinal Salmonella when you look at degradation of the central anaerobic metabolism genes.
Main
I’m interested in identifying pseudogenes (also, and maybe more properly, known as hypothetically disrupted coding sequences [HDCs]) in some Salmonella genomes.
There are two parts to this post, the “of general interest” part, which is an assessment of which tools work best for identifying pseudogenes, and a second more niche part.
I tried three pieces of software:
- bakta annotation
- pseudofinder (with three databases)
- a salmonella pan-genome from uniprot (generated with this search query).
- the bakta full annotation database
- a database of just genome of the specific organism being investigated
- delta bit score, which I got working in this docker image.
- I used a higher threshold for calling positives (97.5 percetnile) than the default (95th percentile of least dispersed side of distribution).
I used the dataset from Nuccio & Bäumler, 2014 as a truth dataset, as these genomes were manually curated. I ran the genomes that were included in that analysis through the various tools/databases, and then judged the results with the Nuccio calls as “truth”. The Nuccio and Bäumler set contains a mix of “extraintestinal” and “gastrointestinal” Salmonella genomes, extra-intestinal Salmonella are known to have a much higher number of pseudogenes than gastrointestinal.
First part – PPV and sensitivity when matched by co-ordinate.
This section relies upon the fact that we can perfectly match the calls back to the truth based on the genomic co-ordinates, as they are the same genomes.
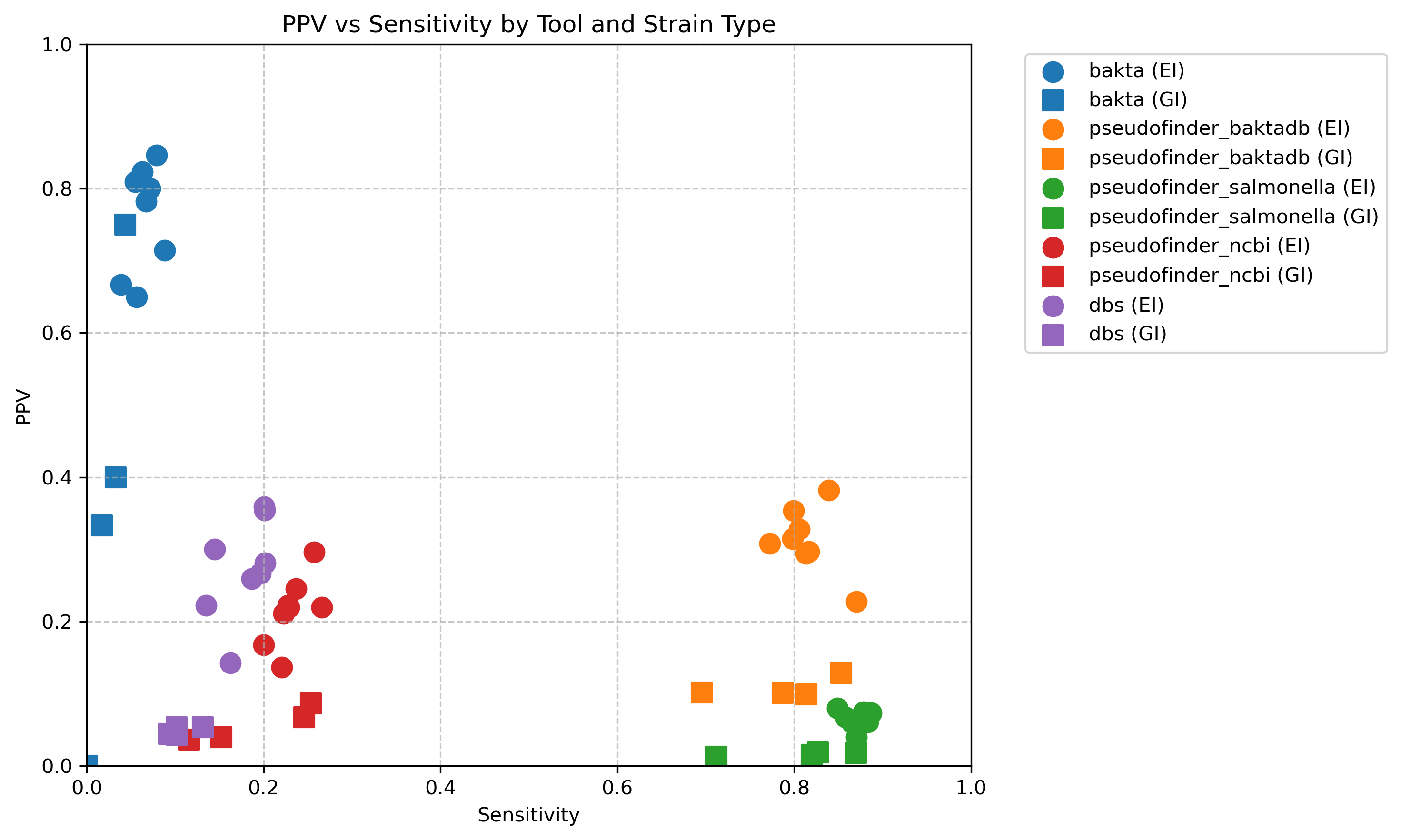
Here’s a scatter plot of the positive predictive value compared with the sensitivity of each approach. Shape is whether it is extraintestinal (EI) or gastrointestinal (GI).
How about the relationship between the true number of pseudogenes and the predicted?
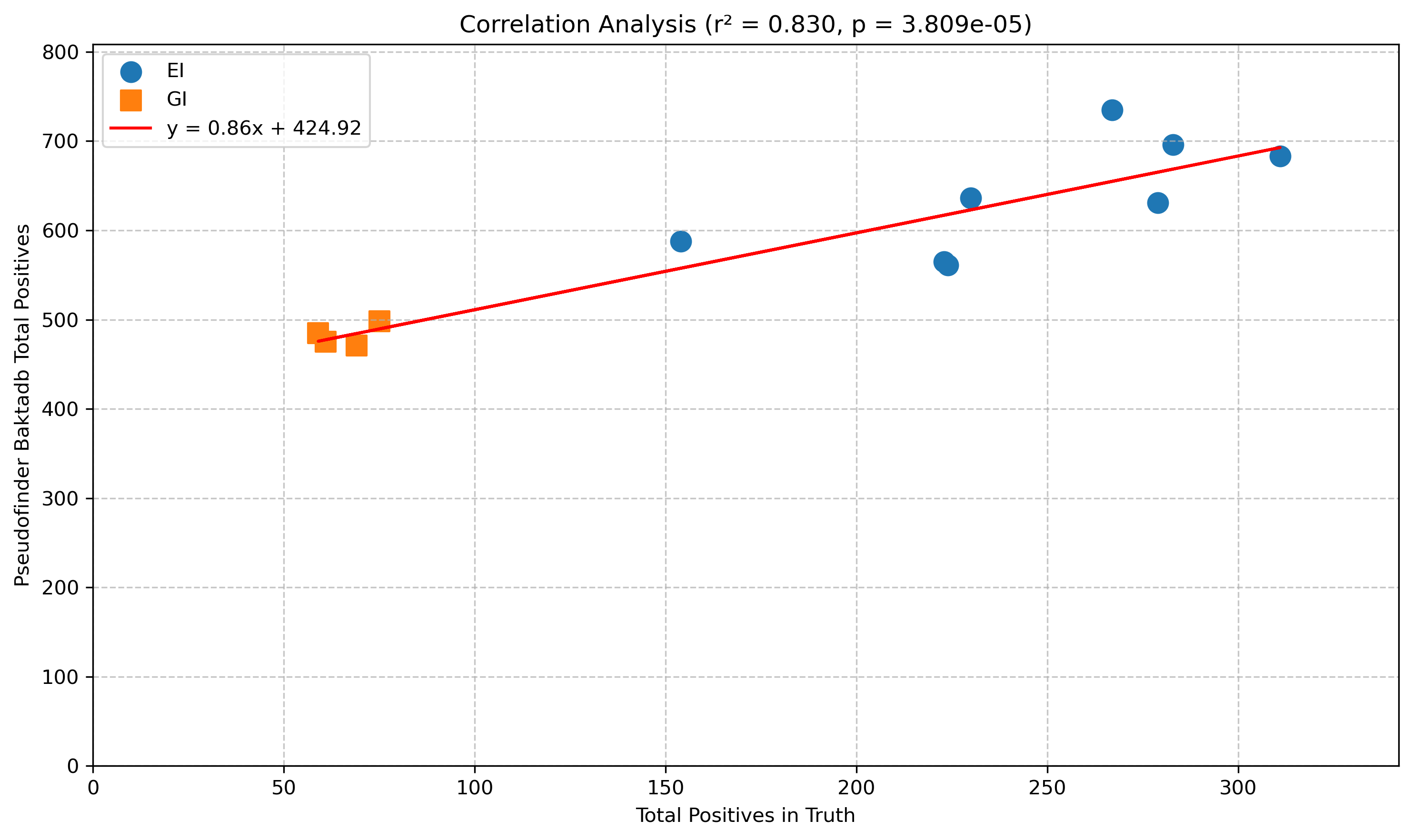
There’s a strong correlation between predicted and true, although with a significant baseline of false positives (around 424 on average). Still, you can broadly trust “more or less” judgements from pseudofinder with bakta db when it comes to counting Salmonella pseudogenes.
Part 2 – PPV and sensitivity when matched by diamond.
The second part of the post is more niche, and applies to my specific question about how these tools do at identifying pseudogenes in a specific biological pathway. The genome I’m interested in was not one of the ones in teh truth set, and so to ensure a fair comparison between the genomes in the truth set and my genome of interest, I had to put them all through the same process. In this case, that was matching the proteins from the genome of interest agains the reference proteome using diamond.
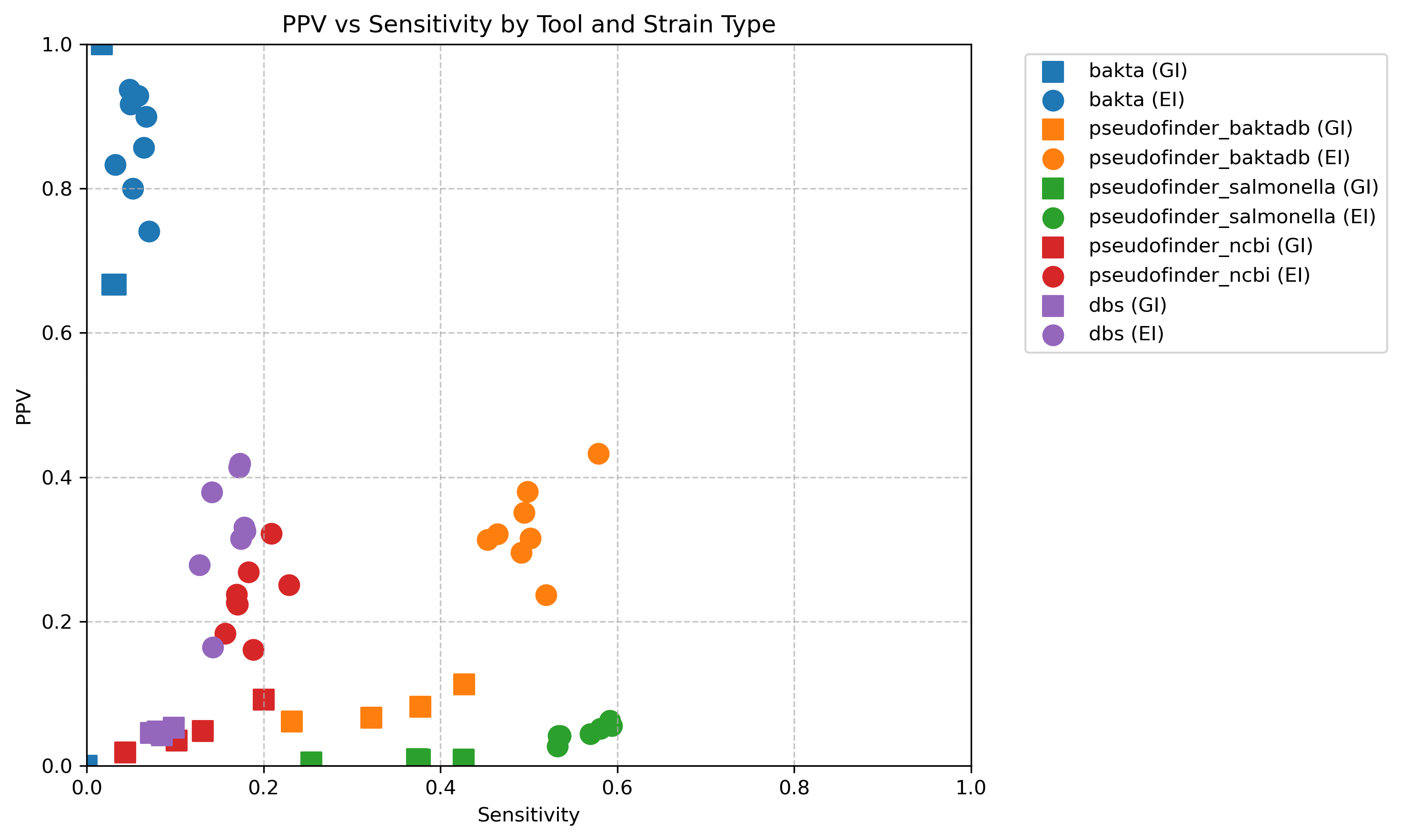
Sensitivity is lower because you have to go through the step of matching the proteins from the genome of interest against the reference proteome by diamond, which inevitably leads to some loss. Perhaps especially of pseudogenes (?), maybe better to work in nucleotide spade for this?
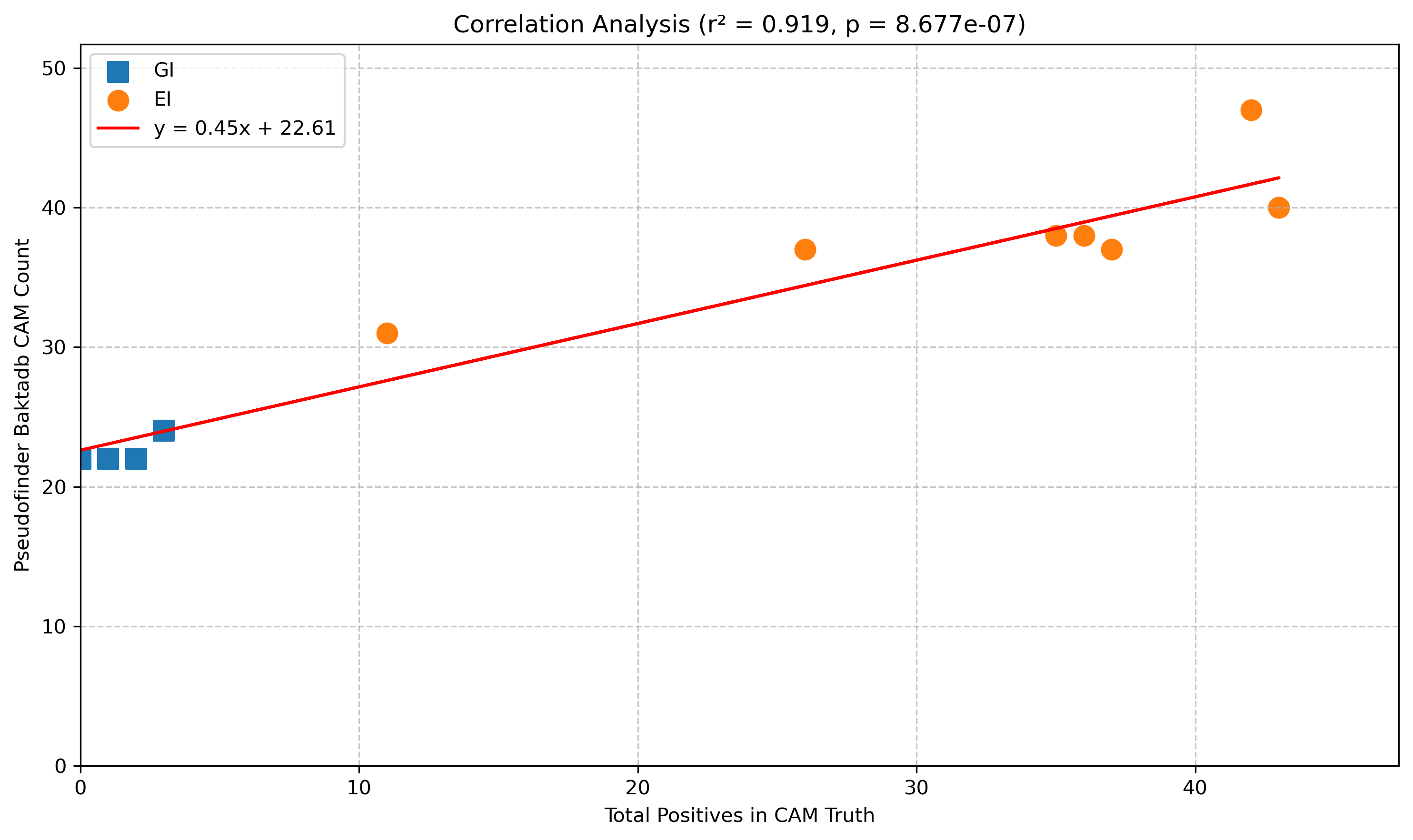
And, for the small number of people who are also interested in pseudogenes in Salmonella central anaerobic metabolism, pseudofinder with baktadb is your best option. There are quite a few false positives, but the key thing here is the separation between the GI and the EI bugs. I.e. if you run this workflow, and your bug has more than 31 pseudogenes in the central anaerobic metabolism genes, it looks more like an EI bug, and if it was less the 24, then it looks more like a GI bug.
Here is the correlation between the number of true pseudogenes in central anaerobic metabolism with the number of pseudofinder identified pseudogenes in central anaerobic metabolism. Strong correlation with a large “baseline” of false positives.
Caveats
This has been quite a CDS/protein focussed approach to validating HDCs/pseudogenes, but this probably isn’t the best approach to this problem, and might be better to work in nucleotide space instead.
Final thoughts/waffle
Just to re-iterate, the difference between the two parts is that in the first, all the calls can be assessed against the truth dataset, while in the second part, only the calls that can be matched against a protein in the truth dataset using diamond can be assessed. If you just have a genome and want to identify pseudo-genes, pay attention to the first part.
I like the elegance of the deltaBS approach, and it would be good to play around with it a bit more, using different databases etc, to see if it’s results can be improved.